
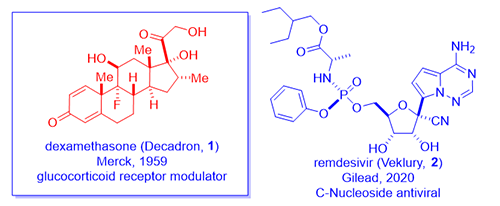
On June 16th, 2020, a team at Oxford University announced that their RECOVERY trials had revealed that a daily treatment of 6 mg of dexamethasone (Decadron, 1) lowered the fatality rate of ventilated COVID-19 patients by up to one third! This was a randomized, double-blinded clinical trial, the gold standard, thus lending much credibility to the results. It seems to work even better than Gilead’s remdesivir (Veklury, 2) for this group of seriously ill patients. Moreover, dexamethasone (1) is cheap, widely available, and it has been used for more sixty years in the clinics.
Let us take a journey to look at how dexamethasone (1) was discovered, how it has become almost a panacea for all ills in medical practice, how it works to benefit COVID-19 patients, and how it is made.
A group of chemists at Merck discovered dexamethasone (1), a steroid hormone, in 1958 as an anti-inflammatory steroid.1 Merck marketed dexamethasone (1) with a brand name Decadron in 1959. As a testimony to how competitive the field was during the golden age of steroid hormone drugs, Schering Corporation simultaneously synthesized the same compound as well. Considering the two companies merged in 2009, it matters not today with regard to priority.
Inflammation and immunity, like all other normal reactions of the body, are meant to preserve or restore health. Classic inflammatory diseases include rheumatoid arthritis and Crohn’s disease (an inflammatory bowel disease). Rheumatoid arthritis is a chronic inflammatory disease characterized by pain, swelling, and subsequent destruction of joints. Philip S. Hench and Edward C. Kendall at the Mayo Clinic discovered cortisone (3) and isolated it from bovine adrenal cortex (a small organ attached to the top of the kidney, cortex means out-layer). In 1948, Hench gave cortisone (3) to desperately ill 29-year-old Mrs. Gardner with severe rheumatoid arthritis. After three days of injections, the long bedridden patient miraculously recovered. She even went downtown and had a three-hour shopping spree! That single event ushered the cortisone era.
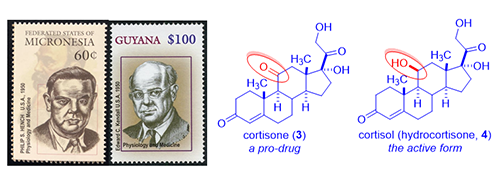
As the understanding of the functions of cortisone (3) increased exponentially, it was soon realized that cortisone (3) itself is a pro-drug, which is reduced in vivo to cortisol (hydrocortisone, 4). Cortisol (4), the actual active drug, is vital to the body’s defense against inflammation. Since Addison’s disease is due to adrenal cortex deficiency and is characterized by the failure of the adrenal glands and the inability to produce cortisol (4), cortisol (4) became the drug of choice for Addison’s disease. President John F. Kennedy suffered from Addison’s disease and was routinely given cortisol (4) shots.
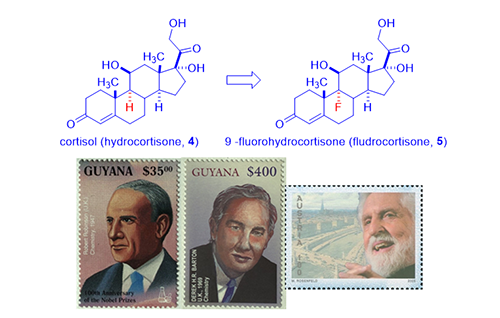
Since steroid hormones cortisone (3) and cortisol (4) were isolated from cortex, they were named as corticosteroids. There are two main groups of corticosteroids, glucocorticoids (good) and mineralocorticoids (bad). While cortisone (3) and cortisol (4) revolutionized medicines, they are far from perfect with many adverse effects, chief among them is mineralocorticoid activity, i.e., salt and water-retaining activity. They are also plagued by metabolic side effects such as weight gain or increased blood glucose levels. Initially, scientists were afraid of tweaking their structures because “nature knows the best”. Fried and Sabo at Squibb in 1954 debunked that notion by preparing semi-synthetic cortisone derivatives with different pharmacological profiles. For instance, their 9a-fluorohydrocortisone (fludrocortisone, 5) was 10-fold more potent than cortisone (3) in relieving rheumatoid arthritis (good) although their mineralocorticoid activity was increased by 300–800-fold (bad, causing aldosterone-like salt and water retaining).3 Since Fried and Sabo showed the world that modifying natural corticosteroids could lead to different, or even better, drugs, chemists in drug industry became bona fide medicinal chemists: they began to explore structure–activity relationship (SAR). More excitingly, almost all of the best organic chemists joined the fray, many of them future Nobel laureates: R. B. Woodward, Robert Robinson, Derek Barton, Carl Djerassi, et al.
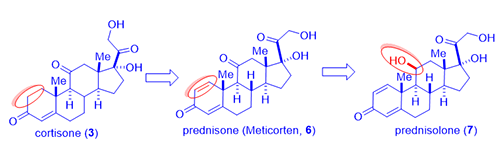
In 1954, from fermentation of cortisone (3) in a mold broth Corynebacterium simplex, Schering Corporation isolated two novel steroids prednisone (C-11 ketone, brand name Meticorten, 6) and the active metabolite prednisolone (C-11 beta hydroxyl, 7) as a consequence of microbial dehydrogenation. These analogs exhibited enhanced glucocorticoid activity with reduced mineralocorticoid activity. The first dose of prednisolone (7) was administered to an arthritic woman in August 1954. Prednisone (6) was launched in March 1955 at a 5-mg dose for use in arthritis, with prednisolone (7) being introduced shortly thereafter. Schering’s initial $100K investment earned them $20 million in the first year of sales, a 200-fold profit.
Both the prodrug prednisone (6) and the active drug prednisolone (7) are approximately five-fold more active than cortisol (hydrocortisone, 4) with less tendency to cause salt retention.
At the time, synthesis of steroids, large molecules with many chiral centers, was long and formidable. Medicinal chemists had no rational design guidelines to follow and progress was dictated by stepwise improvements enabled by synthetic chemistry.4
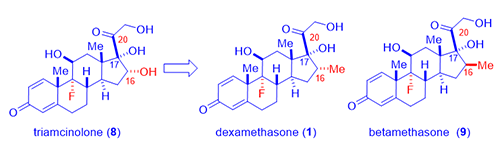
As showcased by fludrocortisone (5), halogen, especially fluorine, incorporation resulted in surprisingly active steroids although fludrocortisone (5) had increased undesirable mineralocorticoid activity. Lederle Laboratories isolated a corticosteroid from the urine sample of a boy taking cortisone (3). The compound had a C-16 hydroxyl group added to the cortisone molecule, no doubt from CYP450 metabolic oxidation. Merging the hydroxyl group and the fluorine substitution on fludrocortisone (5), Lederle arrived at a hybrid molecule, triamcinolone (8), in 1958. The new drug triamcinolone (8) was as potent as prednisolone (7) but was almost free from mineralocorticoid activity. It is not perfect with a tendency to cause nausea, dizziness, and other untoward effects.
In an attempt to hinder metabolism of the side chain corticosteroids (the ketone at the C-20 position was prone to reduction thus loosing activity), Merck chemists led by Sarett synthesized analogs with a methyl group at the adjacent C-16 position such as dexamethasone (1). As often the case in science, the 16-methyl did not really retard metabolism as they initially intended since CYP450 enzymes oxidatively metabolize methyl group with ease. However, unexpectedly, dexamethasone (1) not only acted like a hydroxyl group to depress mineralocorticoid activity, but it also enhance the anti-inflammatory activity by six-fold in comparison to prednisone (6) and 25-times more potent than hydrocortisone (4).1 Dexamethasone (1) has a duration of action of 36–54 hours whereas that of hydrocortisone (4) being 8–12 hours.
Shortly after, Merck also synthesized betamethasone (9), its diastereomer with the methyl group at C-16 position with opposite stereochemistry. Betamethasone (9), a twin brother of dexamethasone (1) in a true sense, behaves nearly identical to each other in terms of biological activities. Apparently, the stereochemistry of the C-16 methyl has little pharmacological consequences. More favorably, both dexamethasone (1) and betamethasone (9) have practically no salt-retaining activity at all. Neither of them exhibited the unwelcome side effects seen with triamcinolone (8).
Today dexamethasone (1) is available as IV, oral, nasal, ophthalmic, and topical cream formulation for treatment of diseases ranging from multiple myeloma to psoriasis. Although glucocorticoids are associated with osteoporosis, fluid retention, and hyperglycemia with chronic therapy, more acute therapy is remarkably efficacious and explains widespread use of steroidal therapy. Unfortunately, the initial enthusiasm for corticosteroids was dampened by their severe side effects following chronic administration. Notably osteoporosis, immune suppression, ulcerogenicity, adrenal suppression, and development of steroid dependence. Prednisone (6) saves lives but takes an awful toll due to side effects, most severe being weakening of the bones within months. As a consequence, for the treatment of inflammatory diseases, corticosteroids have now been largely replaced by non-steroidal anti-inflammatory drugs (NSAIDS). Still, the fact remains that corticosteroids are the most effective drug in the treatment of acute and chronic inflammatory diseases nowadays.
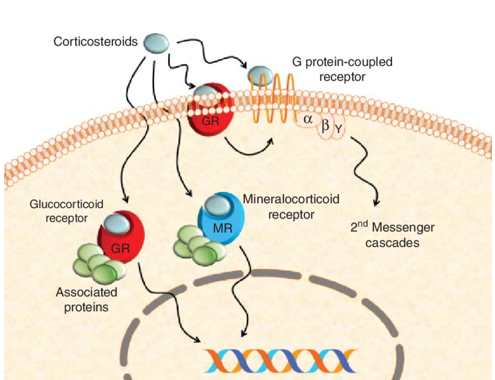
In terms of mechanism of action, dexamethasone (1), like most corticosteroids, is very complicated, hitting multiple drug targets. But its key anti-inflammatory properties are the consequence of serving as a modulator (agonist) of ligand-bound glucocorticoid receptor. As shown in the scheme below, dexamethasone (1) binds to glucocorticoid receptor on cell membrane. The dexamethasone (1)–glucocorticoid receptor complex then enters cell nucleus to interact with nuclear DNA molecules that trigger the signaling pathway for anti-inflammatory outcome. Unfortunately, dexamethasone (1)’s poly-pharmacology comes from its binding to several other targets, chief among them the mineralocorticoid receptor, which is responsible for mineralocorticoid side effects including water and salt-retention.
Since its emergence on the market in 1959, dexamethasone (1) has become one of the two most popular orally bioavailable corticosteroids. The other one is prednisone (6). Having been on the market for over sixty years, dexamethasone (1) has been tried with certain degree of success for many indications, making it seem like a panacea. For instance, it has been found to have efficacy for rheumatic problems, a number of skin diseases, severe allergies, asthma, chronic obstructive lung disease, croup, brain swelling, and along with antibiotics in tuberculosis. Doctors are very familiar with dexamethasone (1)’s pharmacological properties. Therefore, they have been using this drug on severe COVID patients “off label” to relieve their pneumonia symptoms. The RECOVERY clinical trial simply confirmed what the doctor inferred early on at the peak of the pandemic, now they can starting using it with much more confidence.
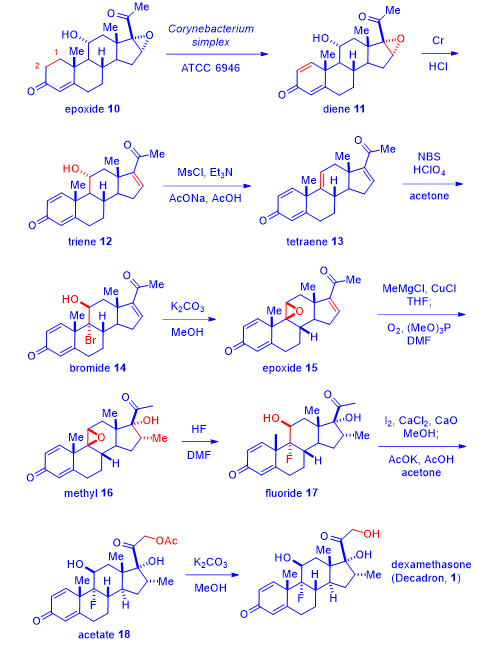
Merck’s initial synthesis of dexamethasone (1) was considered a triumph of steroid chemistry. Today, synthesis of dexamethasone (1) is routine and uneventful. Here is one of the latest version of its synthesis.5
Treatment of commercially available 11a-hydroxyl-16-epoxy progesterone (epoxide 10) with the enzymatic extract of Corynebacterium simplex accomplished the 1,2-deoxygenation to afford diene 11 in the form of a quinone. The efficient microbial biotransformation 10 → 11 was harder to do chemically in a single step. Upon exposure to chromium, the epoxide was reduced to give triene 12. Mesylation and concurrent elimination led to tetraene 13. Bromination of tetraene 13 to afford bromide 14, was chemoselective because the other three olefins were actually enones thus not as reactive. Exposing bromide 14 to basic conditions collapsed the bromohydrin to give epoxide 15. Conjugate addition of the methyl cuprate to compound 15 generated the enolate intermediate, which was oxidized in situ with oxygen to produce methyl compound 16. The epoxide on intermediate 16 was opened by HF to afford fluoride 17. The methyl ketone moiety on fluoride 17 was iodinated and subsequently converted to acetate 18, which was eventually hydrolyzed to deliver dexamethasone (1).5
In summary, dexamethasone (1) has had a colorful “career” as a drug. Initially discovered as an anti-inflammatory drug, it has emerged as one of the two orally bioavailable corticosteroid medicines. It has been used to combat nausea associated with chemotherapy, as an anesthetic, and an anti-allergic. The drug has also found utility to treat asthma, and chronic obstructive pulmonary disease (COPD). Now as an effective medicine to reduce the fatality rate of ventilated COVID-19 patients, dexamethasone (1) continues to save human lives.
References
- Arth, G. E.; Fried, J.; Johnston, D. B. R.; Hoff, D. R.; Sarett, L. H.; Silber, R. H.; Stoerk, H. C.; Winter, C. A. Am. Chem. Soc. 1958, 80, 3161–3163.
- Fried, J.; Sabo, E. F. Am. Chem. Soc. 1954, 76, 1455–1456.
- Hirschmann, R. Steroids 1992, 57, 579–
- Herzog, H.; Oliveto, E. P. Steroids 1992, 57, 617–
- Herraiz, I. Methods Mol. Biol. 2017, 1645, 15–


